Disease-related transcriptomics¶
This page contains descriptions and examples to extract GWAS-implicated gene expression data and project them to cortical and subcortical surfaces. In the following tutorial, we will use epilepsy-related genes (more specifically, genes related to hippocampal sclerosis) as an example, but feel free to replace epilepsy with any other disorder listed below.
Visualize disease-related gene expression maps¶
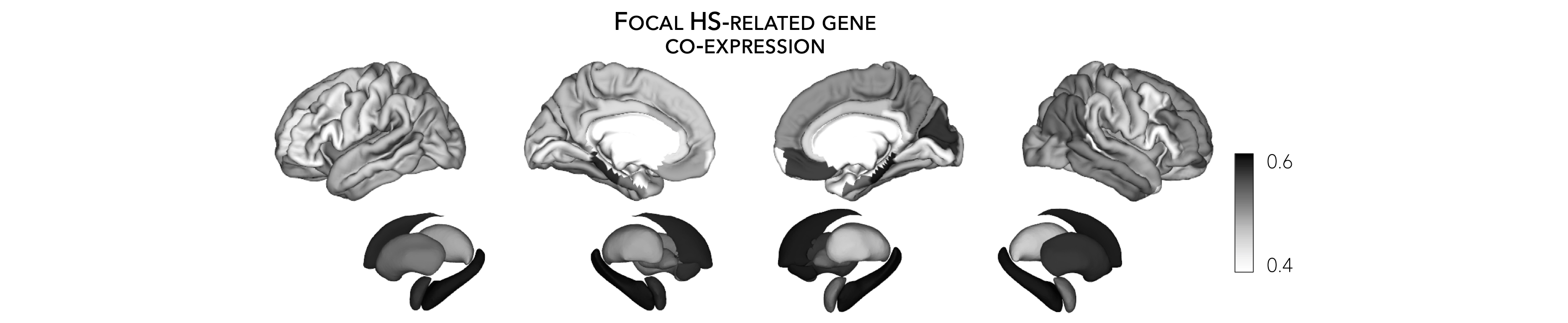
Following up on the above example, we provide a brief example to project gene expression maps to the surface. Once again, we use genes related to hippocampal sclerosis as an example.
Prerequisites ↪ Fetch gene expression data ↪ Extract disease-related gene data
>>> import numpy as np
>>> from enigmatoolbox.utils.parcellation import parcel_to_surface
>>> from enigmatoolbox.plotting import plot_cortical, plot_subcortical
>>> # Compute the mean co-expression across all Focal HS genes
>>> mean_epilepsy_genes = np.mean(epilepsy_gene_data, axis=1)
>>> # Separate cortical (ctx) from subcortical (sctx) regions
>>> mean_epilepsy_genes_ctx = mean_epilepsy_genes[:68]
>>> mean_epilepsy_genes_sctx = mean_epilepsy_genes[68:]
>>> # Map the parcellated gene expression data to our surface template (cortical values only)
>>> mean_epilepsy_genes_ctx_fsa5 = parcel_to_surface(mean_epilepsy_genes_ctx, 'aparc_fsa5')
>>> # Project the results on the surface brain
>>> plot_cortical(array_name=mean_epilepsy_genes_ctx_fsa5, surface_name="fsa5", size=(800, 400), nan_color=(1, 1, 1, 1),
... cmap='Greys', color_bar=True, color_range=(0.4, 0.6))
>>> plot_subcortical(array_name=mean_epilepsy_genes_sctx, ventricles=False, size=(800, 400),
... cmap='Greys', color_bar=True, color_range=(0.4, 0.6))
% Compute the mean co-expression across all Focal HS genes
mean_epilepsy_genes = mean(epilepsy_gene_data{:, :}, 2);
% Separate cortical (ctx) from subcortical (sctx) regions
mean_epilepsy_genes_ctx = mean_epilepsy_genes(1:68);
mean_epilepsy_genes_sctx = mean_epilepsy_genes(69:end);
% Map the parcellated gene expression data to our surface template (cortical values only)
mean_epilepsy_genes_ctx_fsa5 = parcel_to_surface(mean_epilepsy_genes_ctx, 'aparc_fsa5');
% Project the results on the surface brain
f = figure,
plot_cortical(mean_epilepsy_genes_ctx_fsa5, 'color_range', ...
[0.4 0.6], 'cmap', 'Greys')
f = figure,
plot_subcortical(mean_epilepsy_genes_sctx, 'ventricles', 'False', ...
'color_range', [0.4 0.6], 'cmap', 'Greys')